Pipeline Parameters
This page describes all customizable parameters in the MAD4HATTER pipeline, organized by module.
Cutadapt Parameters
Cutadapt handles adapter removal, primer trimming, and quality filtering. These parameters control the demultiplexing and quality control steps.
| Parameter | Description | Default | When to Adjust |
|---|---|---|---|
--cutadapt_minlen | Minimum read length after trimming (shorter reads are discarded) | 100 | Increase if you have short amplicons; decrease to keep more reads |
--quality_score | Quality score threshold for trimming | 20 | Lower (e.g., 15) to keep more reads; higher (e.g., 25) for stricter filtering |
--gtrim | Enable NextSeq-specific quality trimming | false | Set to true if seeing issues with polyG tails (uses --nextseq-trim instead of -q) |
--allowed_errors | Number of mismatches allowed in adapter/primer sequences | 0 | Increase (e.g., 1-2) if primers have known mismatches |
Quality Trimming Behavior
- When
--gtrim false(default): Uses standard quality trimming with-qflag - When
--gtrim true: Uses NextSeq-specific trimming with--nextseq-trimflag
Both use the --quality_score value as the threshold.
!!! warning “Quality Trimming Behavior” --nextseq-trim performs standard quality trimming, and also trims any trailing G tails in the read. This is a common issue when using two colour instruments (e.g., nextseq). However, we found that real biological G bases were being removed by this filter in mad4hatter data. After testing, we found that these poly G tails were being filtered out elsewhere in the pipeline. If you see issues in your data that could be attributed to poly G tails, then you can apply this filtering using the --gtrim flag, however you should be careful to look for unintended consequences.
Example: Custom Cutadapt Parameters
nextflow run main.nf \
--readDIR /path/to/data \
--pools D1,R1,R2 \
--sequencer nextseq \
--cutadapt_minlen 75 \
--quality_score 15 \
--gtrim true \
--allowed_errors 1 \
-profile docker
For more information about Cutadapt parameters, refer to the Cutadapt documentation. —
DADA2 Parameters
DADA2 infers amplicon sequences and can be tuned depending on your needs. These parameters control the sequence inference module.
| Parameter | Description | Default |
|---|---|---|
--omega_a | Abundance threshold - controls whether a sequence is likely a true variant vs. error | 1e-120 |
--dada2_pool | Pooling method for information sharing across samples | pseudo |
--band_size | Alignment heuristic - controls alignment when indels exceed this threshold | 16 |
--maxEE | Maximum expected errors - reads exceeding this are discarded during filtering | 3 |
--maxMismatch | Maximum mismatches allowed in overlap region during read merging | 0 |
--just_concatenate | Concatenate non-overlapping reads instead of discarding | true |
DADA2 Pooling Methods
pseudo(default): Two-round approach that rescues low-abundance allelestrue: Full pooling across all samplesfalse: No pooling - each sample analyzed independently
Pseudo Pooling Details
Pseudo pooling involves:
- First round: DADA2 clustering on individual samples
- Second round: Pooling alleles from all samples as priors, then re-running DADA2
Benefits:
- Rescues low-abundance alleles that appear in multiple samples
- Increases sensitivity for variant detection
Trade-offs:
- Approximately doubles runtime
- May introduce false positives from PCR/sequencing errors
- Default
omega_a(1e-120) helps mitigate false positives
Example: Custom DADA2 Parameters
nextflow run main.nf \
--readDIR /path/to/data \
--pools D1,R1,R2 \
--sequencer nextseq \
--omega_a 1e-100 \
--dada2_pool pseudo \
--band_size 20 \
--maxEE 4 \
--maxMismatch 1 \
-profile docker
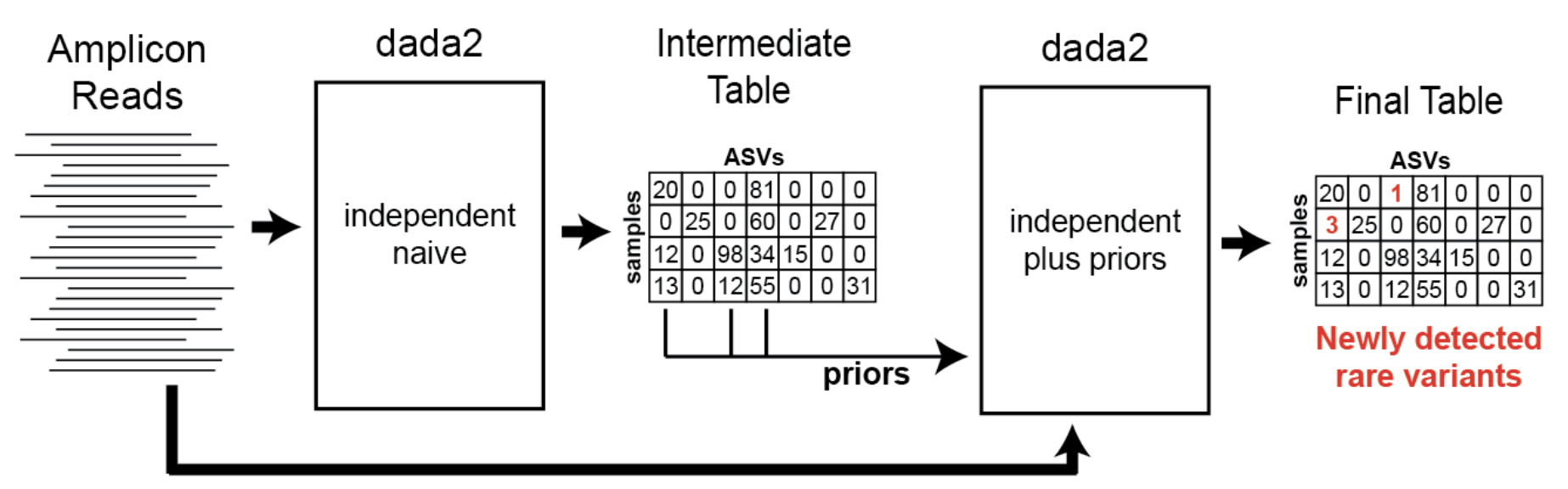 Pseudo-pooling schematic: Benjamin Callahan, https://benjjneb.github.io/dada2/pseudo.html
Pseudo-pooling schematic: Benjamin Callahan, https://benjjneb.github.io/dada2/pseudo.html
For more information about DADA2 parameters, refer to the DADA2 documentation.
Masking Parameters
Control how low-complexity regions (homopolymers and tandem repeats) are masked in sequences.
| Parameter | Description | Default |
|---|---|---|
--mask_homopolymers | Enable homopolymer masking | true |
--homopolymer_threshold | Minimum homopolymer length to mask (e.g., 5 = mask runs of 5+ identical bases) | 5 |
--mask_tandem_repeats | Enable tandem repeat masking | true |
--trf_min_score | Tandem Repeat Finder minimum alignment score | 25 |
--trf_max_period | Tandem Repeat Finder maximum pattern size | 3 |
For more information about Tandem Repeat Finder, refer to the Tandem Repeat Finder documentation
Example: Custom Masking Parameters
nextflow run main.nf \
--readDIR /path/to/data \
--pools D1,R1,R2 \
--sequencer nextseq \
--mask_homopolymers true \
--homopolymer_threshold 3 \
--mask_tandem_repeats true \
--trf_min_score 30 \
-profile docker
Alignment Parameters
Control sequence alignment during post-processing.
| Parameter | Description | Default |
|---|---|---|
--alignment_threshold | Minimum alignment score - sequences below this are filtered out | 60 |
Lower values keep more sequences (including potential off-targets); higher values are more stringent.
Reference Sequence Parameters
Control how reference sequences are provided or generated.
| Parameter | Description | Default |
|---|---|---|
--refseq_fasta | Path to targeted reference sequence file | Auto-generated from pools |
--genome | Path to full genome file (used to generate reference) | Not used |
--amplicon_info | Path to custom amplicon info file | Auto-generated from pools |
!!! note “Reference Priority” If both --refseq_fasta and --genome are provided, --refseq_fasta takes priority. If neither is provided, the pipeline automatically builds a reference from pool configurations.
Resistance Markers
| Parameter | Description | Default | |:———:|:————|:——–| | --resmarker_info | Path to resistance markers of interest covered by the panel | Auto-generated from pools |
!!! Resistance Markers A list of resistance markers of interest is stored in the repository here. The pipeline will check which markers are covered by targets in your panel/pools and report on those. Although this list is extensive, if a marker you care about is missing, you can add it to your local copy of panel_information/principal_resistance_marker_info_table.tsv or provide it in a table via --resmarker_info. If you think the marker would be useful to others, please raise a new issue and label it as a feature request.
Getting Help
To see all available parameters with descriptions:
nextflow run main.nf --help